Motivation¶
shapemapper2 only receives raw read sequences as input, which it then aligns against a target reference sequence using bowtie2 or STAR. As a result, pre-aligned reads must first be converted back to raw read sequences before they can be analysed with shapemapper2—thereby losing some information from their previous alignment.
This can be especially problematic for pre-aligned reads not easily replicable by bowtie2 or STAR alignment through shapemapper2, such as those from splice-aware aligners such as HISAT2, or if the pre-aligned reads were further processed after alignment by programs such as trcls.
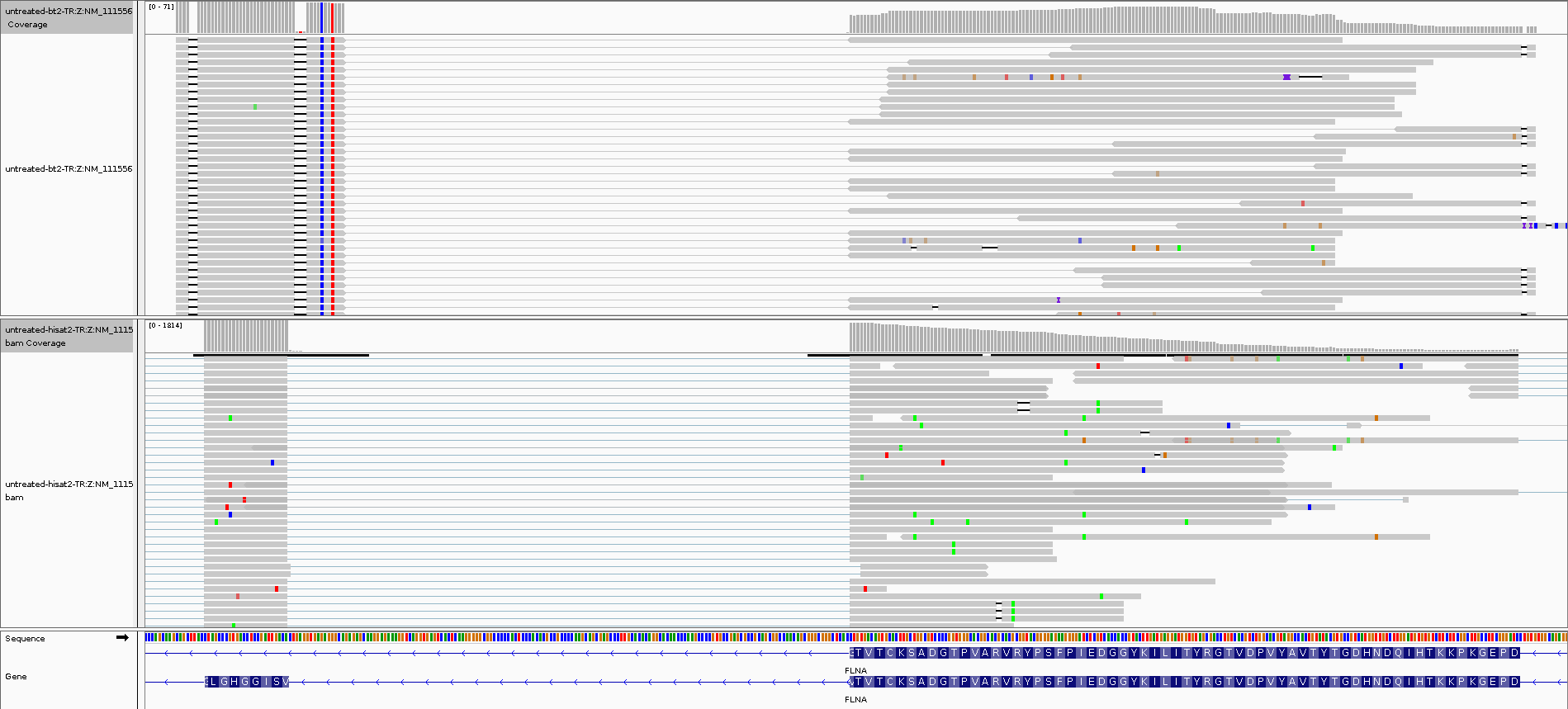
IGV visualisation of a bowtie2 alignment (top) versus a HISAT2 splice-aware alignment (bottom).¶
For transcriptome or genomic alignments, the restriction of the reference region for ‘re-alignment’ in shapemapper2 also causes some reads aligning near the ends of the reference region to fail to align.
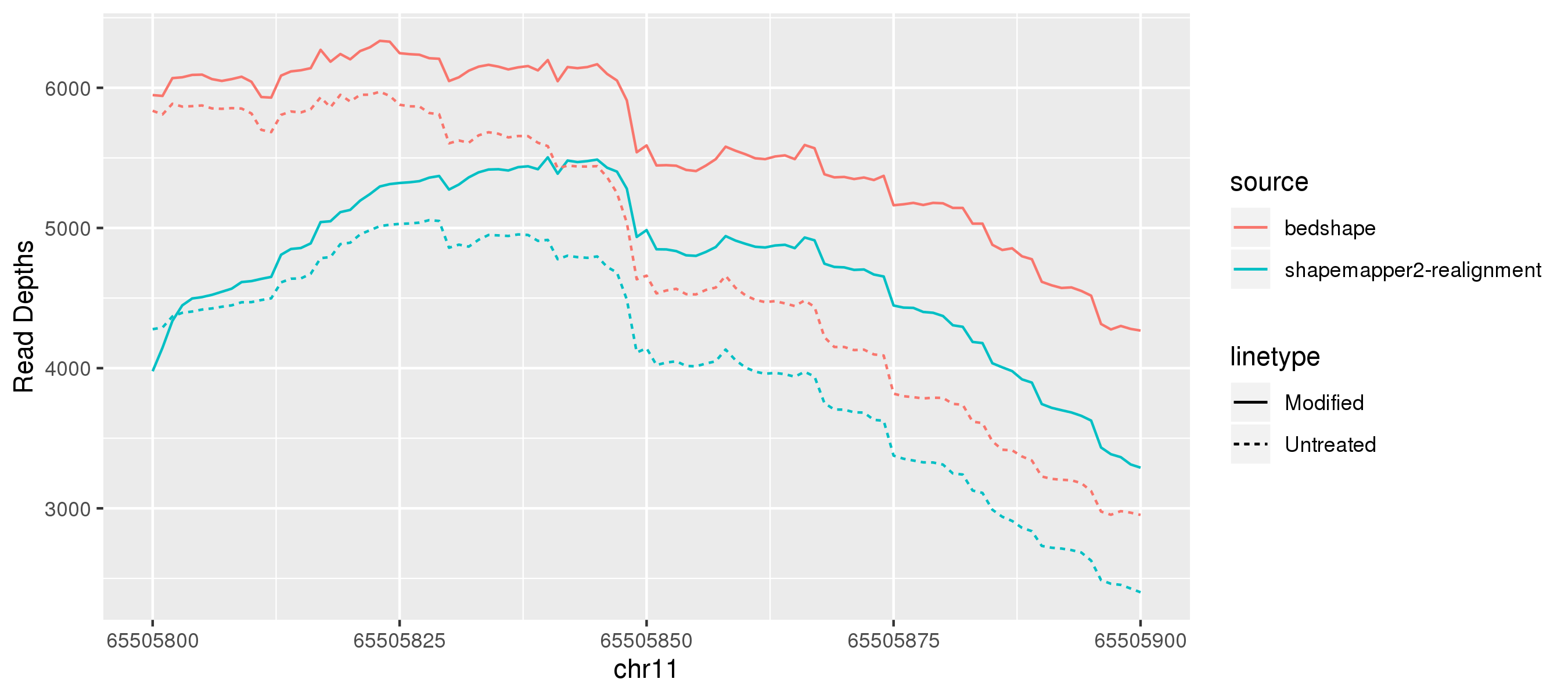
For transcriptome or genomic alignments, re-alignment of already aligned reads against a small reference region causes a proportion of reads to fail to align.¶
bedshape overcomes these problems by soft-clipping each alignment’s position, CIGAR, and MD fields to match the restricted region of interest. Prior alignment information is therefore never completely stripped from the sequence reads—only clipped.
Finally, bedshape provides the functionality to run shapemapper2 reactivity profile calculations for multiple regions in one execution, given a BED file.